Annotation of cell types using Scibet
Florian Wuenenmann
Last updated: 2022-04-19
Checks: 7 0
Knit directory: Abatacept_scrnaseq_mi/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220318) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 6fc473c. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: code/utils.R
Untracked: code/utils_nichenet.R
Untracked: data/Table_1_Common Transcriptomic Effects of Abatacept and Other DMARDs on Rheumatoid Arthritis Synovial Tissue.xlsx
Untracked: omnipathr-log/
Unstaged changes:
Deleted: analysis/7_Conclusions.Rmd
Deleted: analysis/7_summary.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/2_cell_type_annotation.Rmd) and HTML (docs/2_cell_type_annotation.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | a71b19c | Florian Wuennemann | 2022-04-19 | Build site. |
| Rmd | 86ff3e7 | Florian Wuennemann | 2022-04-19 | Main Update to code and adding NichenetR |
| html | 6e3bb2f | Florian Wuennemann | 2022-03-25 | Build site. |
| html | d13934e | Florian Wuennemann | 2022-03-25 | Build site. |
| Rmd | 4bf3cbc | Florian Wuennemann | 2022-03-25 | wflow_publish("analysis/2_cell_type_annotation.Rmd") |
| html | b47d7b0 | Florian Wuennemann | 2022-03-24 | Build site. |
| Rmd | 8591656 | Florian Wuennemann | 2022-03-24 | wflow_publish(c("analysis/1_process_and_integrate_Seurat_Harmony.Rmd", |
| html | 46f2db7 | Florian Wuennemann | 2022-03-23 | First commit. |
Aim
In this analysis, we will use the Seurat object from our first analysis Process and integrate data using Harmony and assign cell type labels, using the annotated Seurat object from Clara’s analysis, as well as pre-processed reference datasets from Scibet. This will help to understand what cell types we have in the data.
Load data
First, let’s load the seurat objects.
## Load Reference and query seurat objects
seurat_object_annotated <- here("../data/2_annotated.seurat_object.rds")
if(!file.exists(seurat_object_annotated)){
seurat_object_filt <- readRDS(here("../data/1_harmony.seurat_object.rds"))
}else{
seurat_object_filt <- readRDS(seurat_object_annotated)
}## Load Reference and query seurat objects
ref_anno <- readRDS(here("../data/CTLA4_annotated.rds"))Transferring cell labels using Scibet
Transfer Clara’s labels
We will use Scibet, a lightweight annotation method in R, to label the Harmony processed Seurat object, using the previously assigned labels from Clara. Since the underlying cells / data is almost exactly the same, this should work quite well.
## Format reference matrix
exp <- t(ref_anno@assays$RNA@data)
cluster_names <- ref_anno@meta.data$cluster.ids
exp <- as_tibble(exp) %>%
mutate("label" = cluster_names)## Format query matrix
exp_query <- t(seurat_object_filt@assays$RNA@data)## Run Scibet
pred_scibet <- SciBet(exp, exp_query)## Assign cell types based on Scibet predictions
seurat_object_filt$cell_type <- pred_scibet## Check whether the annotaiton using Clara's labels worked well
umap_plot_scibet <- DimPlot(seurat_object_filt, reduction = "umap", group.by = "cell_type", label = TRUE)
umap_plot_scibet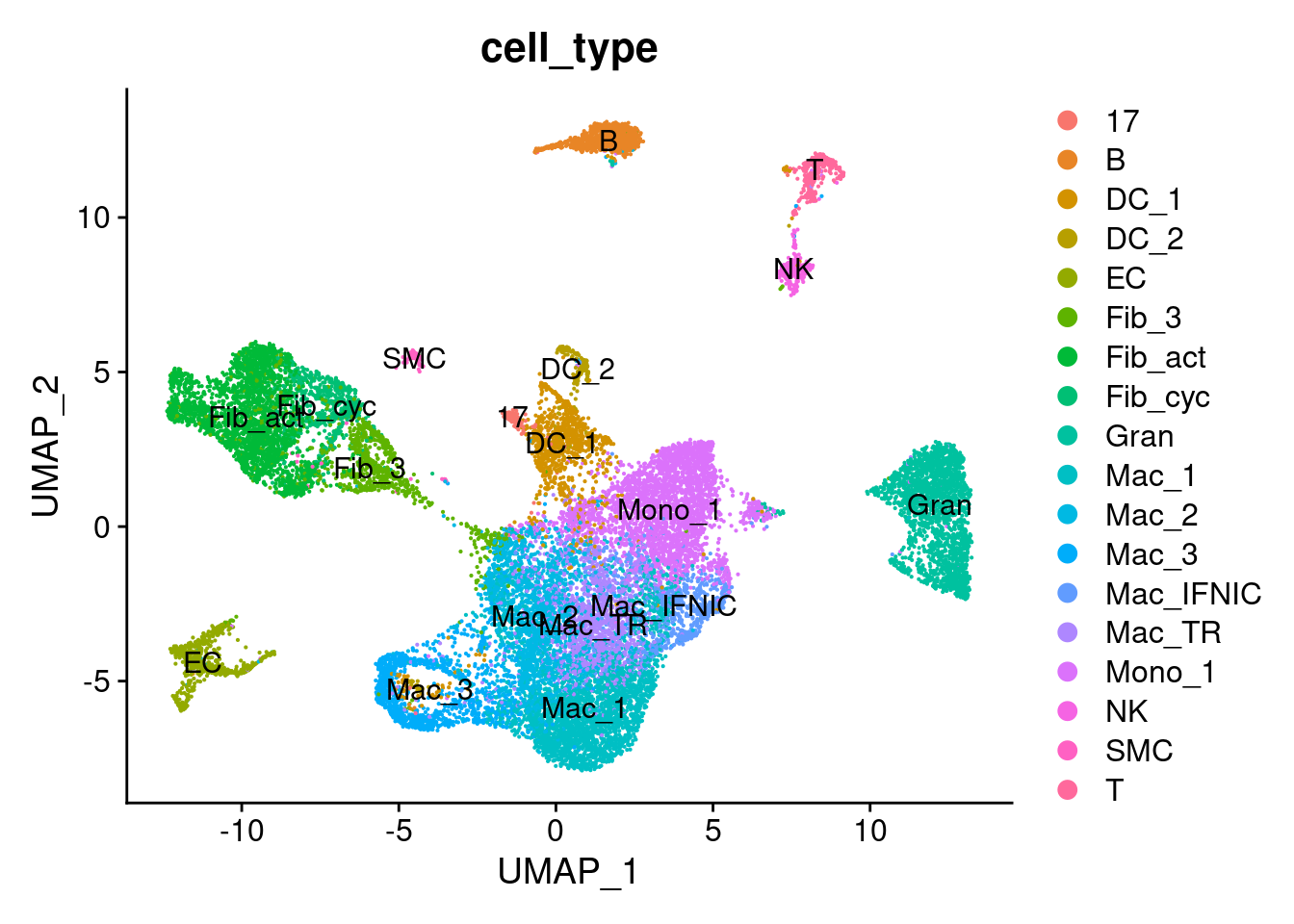
| Version | Author | Date |
|---|---|---|
| a71b19c | Florian Wuennemann | 2022-04-19 |
save_plot(umap_plot_scibet,
filename = here("../results/UMAP.harmony.scibet_anno.png"),
base_height = 5)Using Scibet pre-processed references
Let’s use two different existing annotations from public datasets to label the cells. This might give us an idea of what cluster we are unsure about could be.
Tabula Muris
First, let’s use the popular Tabula Muris dataset to annotate the data. This dataset has a number of different cell types from diverse mouse organs in it and should therefore cover most basic cell types.
tabula_muris_ref <- readr::read_csv(here("../references/GSE109774_scibet_core.csv"))New names:
Rows: 80 Columns: 1001
── Column specification
──────────────────────────────────────────────────────── Delimiter: "," chr
(1): ...1 dbl (1000): Prss34, Sftpc, Hba-a1, Pyy, Sst, Nppa, Ppy, Gcg, Ins1,
Ctrb1, Um...
ℹ Use `spec()` to retrieve the full column specification for this data. ℹ
Specify the column types or set `show_col_types = FALSE` to quiet this message.
• `` -> `...1`model <- pro.core(tabula_muris_ref)
prd <- LoadModel(model)## Run Scibet
pred_scibet_tabulamuris <- prd(exp_query)seurat_object_filt$tabula_muris <- pred_scibet_tabulamuris
umap_plot_scibet_tabula_muris <- DimPlot(seurat_object_filt, reduction = "umap", group.by = "tabula_muris", label = FALSE)
HoverLocator(plot = umap_plot_scibet_tabula_muris, information = FetchData(seurat_object_filt,vars = c("tabula_muris")))Warning in if (is.na(col)) {: the condition has length > 1 and only the first
element will be used
Warning in if (is.na(col)) {: the condition has length > 1 and only the first
element will be usedWarning: `error_y.color` does not currently support multiple values.Warning: `error_x.color` does not currently support multiple values.Warning: `line.color` does not currently support multiple values.Warning: The titlefont attribute is deprecated. Use title = list(font = ...)
instead.## What is cluster 17?
cluster_17_tabulamuris <- seurat_object_filt@meta.data %>%
subset(cell_type == "17") %>%
group_by(tabula_muris) %>%
tally()
cluster_17_tabulamuris# A tibble: 4 × 2
tabula_muris n
<chr> <int>
1 blood cell 2
2 macrophage 1
3 myeloid cell 65
4 professional antigen presenting cell 50Smart-seq2 - Heart and Aorta
heart_aorta <- readr::read_csv(here("../references/GSE109774_Heart_Smart-seq2_scibet_core.csv"))New names:
Rows: 8 Columns: 1001
── Column specification
──────────────────────────────────────────────────────── Delimiter: "," chr
(1): ...1 dbl (1000): Lyz2, Nppa, Acta2, Cd74, H2-Ab1, Ttn, H2-Aa, Dcn, Actc1,
Tnni3, ...
ℹ Use `spec()` to retrieve the full column specification for this data. ℹ
Specify the column types or set `show_col_types = FALSE` to quiet this message.
• `` -> `...1`model <- pro.core(heart_aorta)
prd <- LoadModel(model)## Run Scibet
pred_scibet_heart <- prd(exp_query)seurat_object_filt$smartseq_ha <- pred_scibet_heart
umap_plot_scibet_heart <- DimPlot(seurat_object_filt, reduction = "umap", group.by = "smartseq_ha", label = FALSE)
HoverLocator(plot = umap_plot_scibet_heart, information = FetchData(seurat_object_filt,vars = c("smartseq_ha")))Warning in if (is.na(col)) {: the condition has length > 1 and only the first
element will be used
Warning in if (is.na(col)) {: the condition has length > 1 and only the first
element will be usedWarning: `error_y.color` does not currently support multiple values.Warning: `error_x.color` does not currently support multiple values.Warning: `line.color` does not currently support multiple values.Warning: The titlefont attribute is deprecated. Use title = list(font = ...)
instead.## What is cluster 17?
cluster_17_smartseq_ha <- seurat_object_filt@meta.data %>%
subset(cell_type == "17") %>%
group_by(smartseq_ha) %>%
tally()
cluster_17_smartseq_ha# A tibble: 1 × 2
smartseq_ha n
<chr> <int>
1 leukocyte 118Compare Seurat cluster mappings between R and harmony integration
meta_ref <- ref_anno@meta.data
meta_harmony <- seurat_object_filt@meta.data
rm(ref_anno)meta_ref_sel <- meta_ref %>%
rownames_to_column() %>%
mutate("rowname" = gsub("GEX_","",rowname))
meta_harmony_sel <- meta_harmony %>%
rownames_to_column()
merged_meta <- full_join(meta_ref_sel,meta_harmony_sel, by ="rowname")
## How many cells perfectly overlap clusters
cluster_overlap <- merged_meta %>%
mutate("overlap" = if_else(cluster.ids == cell_type,"yes","no"),
"unique_in" = if_else(is.na(cell_type),"R_integration",
if_else(is.na(cluster.ids),"harmony","both")))
cluster_overlap_num <- cluster_overlap %>%
group_by(overlap) %>%
tally()
cluster_overlap_num# A tibble: 3 × 2
overlap n
<chr> <int>
1 no 2715
2 yes 15112
3 <NA> 7851cluster_overlap_group <- cluster_overlap %>%
group_by(unique_in) %>%
tally()
cluster_overlap_group# A tibble: 3 × 2
unique_in n
<chr> <int>
1 both 17827
2 harmony 3751
3 R_integration 4100Save the seurat object with the full annotations
## Set cell type as new main annotation
Idents(seurat_object_filt) <- "cell_type"if(!file.exists(seurat_object_annotated)){
saveRDS(seurat_object_filt, file = seurat_object_annotated)
}Identify marker genes using
Before transferring labels from Clara’s annotated object, let’s calculate marker genes for the annotated seurat clusters using presto, which is much faster than the default Seurat FindMarker function.
seurat_marker <- wilcoxauc(seurat_object_filt,
group_by = 'seurat_clusters',
assay = "data",
seurat_assay = "RNA")## Lets visualize the top markers in a table
seurat_marker_top <- seurat_marker %>%
group_by(group) %>%
top_n(10, wt = auc) %>%
arrange(desc(auc)) %>%
ungroup()
reactable(seurat_marker_top,
resizable = TRUE, showPageSizeOptions = TRUE,
onClick = "expand", highlight = TRUE)
sessionInfo()R version 4.1.3 (2022-03-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=de_DE.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=de_DE.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=de_DE.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=de_DE.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] here_1.0.1 reactable_0.2.3
[3] presto_1.0.0 data.table_1.14.2
[5] Rcpp_1.0.8.3 cowplot_1.1.1
[7] forcats_0.5.1 stringr_1.4.0
[9] dplyr_1.0.8 purrr_0.3.4
[11] readr_2.1.2 tidyr_1.2.0
[13] tibble_3.1.6 tidyverse_1.3.1
[15] scibet_1.0 SingleR_1.8.1
[17] SummarizedExperiment_1.24.0 Biobase_2.54.0
[19] GenomicRanges_1.46.1 GenomeInfoDb_1.30.1
[21] IRanges_2.28.0 S4Vectors_0.32.4
[23] BiocGenerics_0.40.0 MatrixGenerics_1.7.0
[25] matrixStats_0.61.0 patchwork_1.1.1
[27] ggplot2_3.3.5 SeuratObject_4.0.4
[29] Seurat_4.1.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] utf8_1.2.2 R.utils_2.11.0
[3] reticulate_1.24 tidyselect_1.1.2
[5] htmlwidgets_1.5.4 grid_4.1.3
[7] BiocParallel_1.28.3 Rtsne_0.15
[9] munsell_0.5.0 ScaledMatrix_1.2.0
[11] codetools_0.2-18 ica_1.0-2
[13] future_1.24.0 miniUI_0.1.1.1
[15] withr_2.5.0 spatstat.random_2.2-0
[17] colorspace_2.0-3 highr_0.9
[19] knitr_1.38 rstudioapi_0.13
[21] ROCR_1.0-11 tensor_1.5
[23] listenv_0.8.0 labeling_0.4.2
[25] git2r_0.30.1 GenomeInfoDbData_1.2.7
[27] polyclip_1.10-0 bit64_4.0.5
[29] farver_2.1.0 rprojroot_2.0.3
[31] parallelly_1.31.0 vctrs_0.4.1
[33] generics_0.1.2 xfun_0.30
[35] R6_2.5.1 rsvd_1.0.5
[37] flexmix_2.3-17 bitops_1.0-7
[39] spatstat.utils_2.3-0 DelayedArray_0.20.0
[41] assertthat_0.2.1 vroom_1.5.7
[43] promises_1.2.0.1 scales_1.2.0
[45] nnet_7.3-17 gtable_0.3.0
[47] beachmat_2.10.0 globals_0.14.0
[49] processx_3.5.3 goftest_1.2-3
[51] rlang_1.0.2 splines_4.1.3
[53] lazyeval_0.2.2 spatstat.geom_2.4-0
[55] broom_0.8.0 BiocManager_1.30.16
[57] yaml_2.3.5 reshape2_1.4.4
[59] abind_1.4-5 modelr_0.1.8
[61] crosstalk_1.2.0 backports_1.4.1
[63] httpuv_1.6.5 tools_4.1.3
[65] ellipsis_0.3.2 spatstat.core_2.4-2
[67] jquerylib_0.1.4 RColorBrewer_1.1-3
[69] ggridges_0.5.3 plyr_1.8.7
[71] sparseMatrixStats_1.7.0 zlibbioc_1.40.0
[73] RCurl_1.98-1.6 ps_1.6.0
[75] rpart_4.1.16 deldir_1.0-6
[77] pbapply_1.5-0 zoo_1.8-9
[79] reactR_0.4.4 haven_2.4.3
[81] ggrepel_0.9.1 cluster_2.1.3
[83] fs_1.5.2 magrittr_2.0.3
[85] scattermore_0.8 lmtest_0.9-40
[87] reprex_2.0.1 RANN_2.6.1
[89] whisker_0.4 fitdistrplus_1.1-8
[91] hms_1.1.1 mime_0.12
[93] evaluate_0.15 xtable_1.8-4
[95] readxl_1.4.0 gridExtra_2.3
[97] compiler_4.1.3 KernSmooth_2.23-20
[99] crayon_1.5.1 R.oo_1.24.0
[101] htmltools_0.5.2 mgcv_1.8-40
[103] later_1.3.0 tzdb_0.3.0
[105] lubridate_1.8.0 DBI_1.1.2
[107] dbplyr_2.1.1 MASS_7.3-56
[109] Matrix_1.4-1 cli_3.2.0
[111] R.methodsS3_1.8.1 parallel_4.1.3
[113] igraph_1.3.0 pkgconfig_2.0.3
[115] getPass_0.2-2 plotly_4.10.0
[117] spatstat.sparse_2.1-0 xml2_1.3.3
[119] bslib_0.3.1 XVector_0.34.0
[121] rvest_1.0.2 callr_3.7.0
[123] digest_0.6.29 sctransform_0.3.3
[125] RcppAnnoy_0.0.19 spatstat.data_2.1-4
[127] rmarkdown_2.13 cellranger_1.1.0
[129] leiden_0.3.9 uwot_0.1.11
[131] DelayedMatrixStats_1.16.0 modeltools_0.2-23
[133] shiny_1.7.1 lifecycle_1.0.1
[135] nlme_3.1-157 jsonlite_1.8.0
[137] SeuratWrappers_0.3.0 BiocNeighbors_1.12.0
[139] viridisLite_0.4.0 fansi_1.0.3
[141] pillar_1.7.0 lattice_0.20-45
[143] fastmap_1.1.0 httr_1.4.2
[145] survival_3.2-13 remotes_2.4.2
[147] glue_1.6.2 png_0.1-7
[149] bit_4.0.4 stringi_1.7.6
[151] sass_0.4.1 BiocSingular_1.10.0
[153] irlba_2.3.5 future.apply_1.8.1